4. Combining BioServices with external tools
This section shows how to use BioServices as an intermediate tool that fetch data to be used by third-party software/application.
The external applications used in this section are not part of BioServices therefore we do not provide instructions for the installation. Reader should refer to the application web site instead (URLs are provided here below). However, we indicate the way we installed them.
4.1. PYMOL
This example below uses the external software called PyMOL. It can be installed via conda:
conda install -c schrodinger pymol-bundle
The following code uses BioServices to get the PDB Identifier of a protein
called ZAP70. To do so, we use bioservices.uniprot.UniProt to get its accession number (P43403) and its
PDB identifer. Then, we use bioservices.pdb.PDB to get the 3D structure in PDB
format.
1import __main__
2
3__main__.pymol_argv = ["pymol", "-qc"] # Quiet and no GUI
4
5import os
6
7if os.path.isfile("bioservices_pdb.png"):
8 os.remove("bioservices_pdb.png")
9
10# BioServices 1: obtain the PDB ID from a given uniprot ID (P43403 i.e. ZAP70)
11from bioservices import PDBe, UniProt
12
13print("Retrieving PDB ID")
14u = UniProt(verbose=False)
15res = u.mapping(fr="UniProtKB_AC-ID", to="PDB", query="P43403")
16pdb_id = res["results"][0]["to"] # e.g, "1FBV"
17
18# BioServices 2: Download the PDB file from the PDB Web Service
19print("Fetching PDB file")
20p = PDBe()
21res = p.get_files(pdb_id)
22
23# General: save the fetched file in a temporary file
24import tempfile
25
26fh = tempfile.NamedTemporaryFile()
27fh.write(res)
28sname = fh.name
29
30# THIS IS NOT BIOSERVICES ANYMORE but PYMOL
31import pymol
32
33pymol.finish_launching()
34pymol.cmd.load(sname)
35pymol.cmd.png("bioservices_pdb.png", width="15cm", height="15cm", dpi=140)
36# pymol.cmd.png("my_image.png")
37# Get out!
38pymol.cmd.quit()
The script above uses PyMOL in a script manner to save the 3D graphical representation of the protein (here below) but you could also use PyMOL in an interactive mode.

4.2. BioPython
BioPython provides many tools for IO, algorithms and access to Web services. BioServices provides access to many web services. This example shows how (i) to use BioServices to retrieve FASTA files and (ii) BioPython to play with the sequences.
Note
We assume you have installed BioPython (pip install biopython)
First, let us retrieve two FASTA sequences and save them in 2 files:
from bioservices import UniProt
u = UniProt()
akt1 = u.retrieve("P31749", "fasta")
akt2 = u.retrieve("P31751", "fasta")
fh = open("akt1.fasta", "w")
fh.write(akt1)
fh.close()
fh = open("akt2.fasta", "w")
fh.write(akt2)
fh.close()
Now, on the BioPython side, we read the 2 sequences and introspect them:
>>> from Bio import SeqIO, AlignIO
>>> record1 = SeqIO.read("akt1.fasta", "fasta")
>>> record2 = SeqIO.read("akt2.fasta", "fasta")
>>> record1 += "-" # this is to have 2 sequences on same length as requested by the following function
>>> alignment = AlignIO.MultipleSeqAlignment([])
>>> alignment.append(record1)
>>> alignment.append(record2)
>>> for record in alignment:
>>> print(record.description)
sp|P31749|AKT1_HUMAN RAC-alpha serine/threonine-protein kinase OS=Homo sapiens GN=AKT1 PE=1 SV=2
sp|P31751|AKT2_HUMAN RAC-beta serine/threonine-protein kinase OS=Homo sapiens GN=AKT2 PE=1 SV=2
You are ready to play with BioPython multiple alignment tools. Please consult BioPython documentation for more examples.
4.3. Galaxy
- URL:
- Date:
Aug 2013
Galaxy is an open, web-based platform for accessible, reproducible, and transparent computational biomedical research. It provides worflows and plugins to many web resources.
This tutorial shows how to link bioservices and galaxy. Our tutorial will provide a plugin to Galaxy so that a user can retrieve a FASTA file via BioServices and the wrapping of UniProt Web Services.
We assume that you installed Galaxy on your system via the source code:
git clone https://github.com/galaxyproject/galaxy.git
cd galaxy
The tree directory should therefore contain a directory called tools/ and in the config directory, an XML file called tool_conf.xml
We will first create a plugin for bioservices. This is done by adding a directory called bioservices in ./tools:
mkdir tools/bioservices
In this directory, we will create two files called uniprot.py that will contain the actual code that calls bioservices and a second XML file that will allows us to design the plugin layout.
Let us start with the plugin. It is very simple since only the UniProt Entry is required. The output will simply be the FASTA file that would have been fetched.
The XML file is:
<tool id="bioservices_uniprot" name="Get FASTA" version="1.1.0">
<description>from UniProt via Bioservices</description>
<requirements>
<requirement type="package">bioservices</requirement>
</requirements>
<command interpreter="python">uniprot.py $uniprot_id $output</command>
<inputs>
<param name="uniprot_id" type="text" label="UniProt ID" size="40" help="Provide a valid UniProt Entry (e.g. P43403) "/>
</inputs>
<outputs>
<data format="fasta" name="output" />
</outputs>
<help>
Fetch a FASTA file using UniProt via BioServices. Simply provide a valid Uniprot Entry (e.g., P43403)
</help>
</tool>
The python code will take as an input the UniProt ID and create a file that contains the FASTA data:
import sys
def __main__():
ids = sys.argv[1]
filename = sys.argv[2]
# TODO: check the validity and format ?
try:
from bioservices import UniProt
u = UniProt(verbose=False)
u.debugLevel = "ERROR"
except ImportError:
print("Could not import bioservoces ? Check that it is installed. Try 'pip install bioservices'")
try:
fasta = u.searchUniProtId(ids, "fasta")
except:
print("An error occured while fetching the FASTA file from uniprot")
try:
fh = open(filename, "w")
fh.write(fasta)
Finally, you need to make Galaxy aware of this new plugin. This is done in
config/tool_conf.xml. Add the bioservices plugin entry. The beginning of
the file should look like:
<?xml version="1.0"?>
<toolbox>
<section name="Get Data" id="getext">
<tool file="bioservices/uniprot.xml"/>
<tool file="data_source/upload.xml"/>
...
Once done. start you galaxy server. The following image show the outcome: in the left hand side, you can select the bioservices plugin. Then, in the center, you can enter a uniprot entry. Press the execute button and the new file should appear in the right hand side. From there you can use Galaxy other tools to analyse the file.
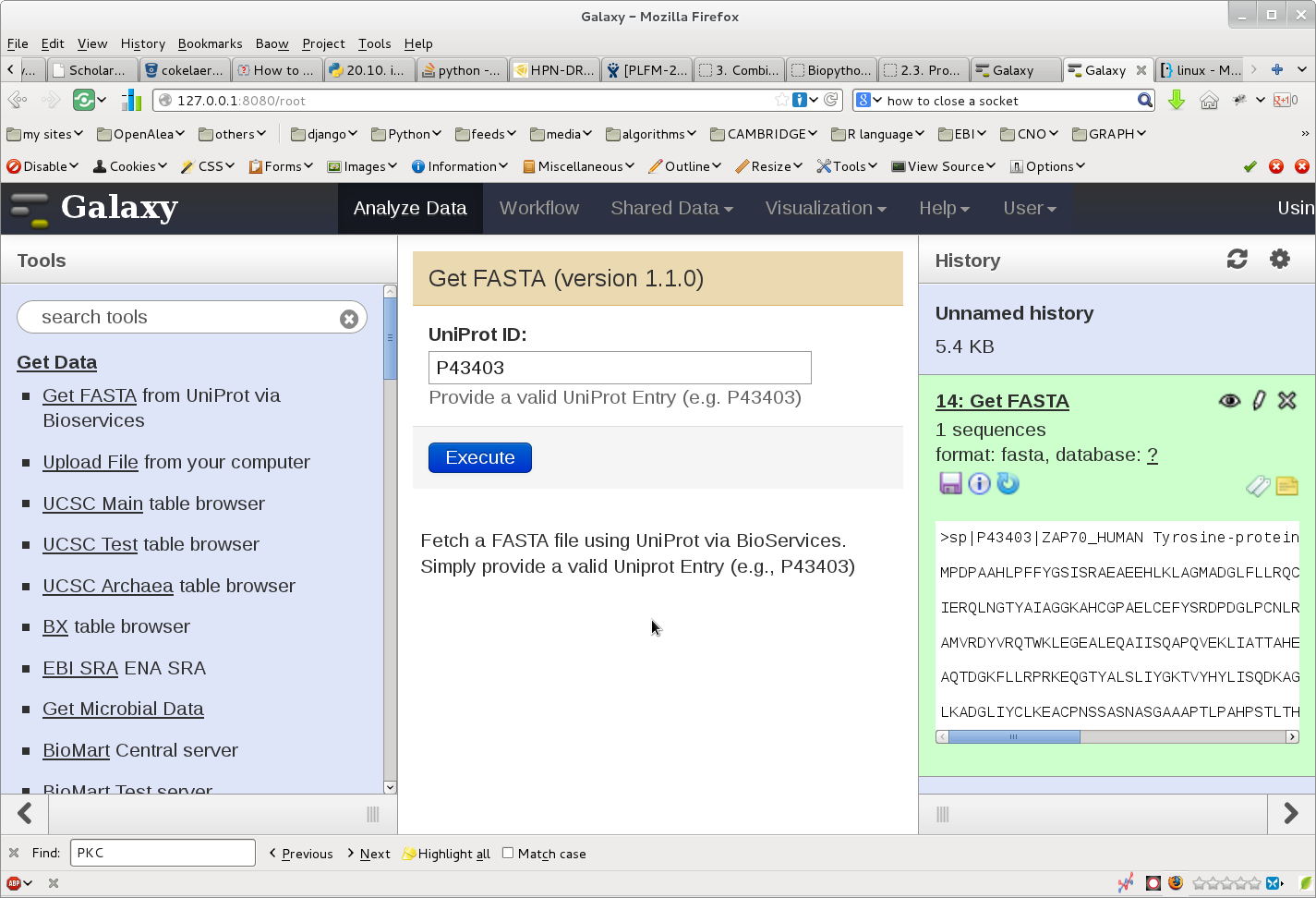
This example shows that it is possible to link Galaxy and BioServices to access to various Web Services that are available through Bioservices.