2.1. KEGG Tutorial
2.1.1. Introduction
Start a kegg interface (default organism is human, that is called hsa):
from bioservices.kegg import KEGG
k = KEGG()
KEGG has many databases. The list can be found in the attribute
bioservices.kegg.KEGG.databases. Each database can be
queried with the bioservices.kegg.KEGG.list() method:
k.list("organism")
The output contains Id of the organism and some other information. To retrieve the Ids, you will need to process the output. However, we provide an alias:
print(k.organismIds)
In general, methods require an access to the on-line KEGG database therefore it takes time. For instance, the command above takes a couple of seconds. However, some are buffered so next time you call it, it will be much faster.
Another useful alias is the pathwayIds to retrieve all pathway Ids. However, you must first specify the organism you are interested in. From the command above we know that hsa (human) is valid organism Id, so let us set it and then get the list of pathways:
k.organism = "hsa"
k.pathwayIds
Another function provided by the KEGG API is the
bioservices.kegg.KEGG.get() one that query a specific entry. Here we are
interested into the human gene with the code 7535:
k.get("hsa:7535") #hsa:7535 is also known as ZAP70
It is quite verbose and is a single string, which may be tricky to handle. We
provide a tool to ease the parsing (see below and bioservices.kegg.KEGG.parse()) returned by bioservices.kegg.KEGG.parse().
2.1.2. Searching for an organism
The method bioservices.kegg.KEGG.find() is quite convenient to search for
entries in different database. For instance, if you want to know the code of
an entry for the gene called ZAP70 in the human organism, type:
>>> s.find("hsa", "zap70")
'hsa:7535\tZAP70, SRK, STCD, STD, TZK, ZAP-70; zeta-chain (TCR) associated protein kinase 70kDa (EC:2.7.10.2); K07360 tyrosine-protein kinase ZAP-70 [EC:2.7.10.2]\n'
It is quite powerful and more examples will be shown. However, it has some limitations.
For example, what about searching for the organism Ids that correspond to any
Drosophila? It does not look like it is possible. BioServices provides a method to search
for an organism Id using lookfor_organism() given
the name (or part of it):
>>> k.lookfor_organism("droso")
['T00030 dme Drosophila melanogaster (fruit fly) Eukaryotes;Animals;Arthropods;Insects',
'T01032 dpo Drosophila pseudoobscura pseudoobscura Eukaryotes;Animals;Arthropods;Insects',
'T01059 dan Drosophila ananassae Eukaryotes;Animals;Arthropods;Insects',
'T01060 der Drosophila erecta Eukaryotes;Animals;Arthropods;Insects',
'T01063 dpe Drosophila persimilis Eukaryotes;Animals;Arthropods;Insects',
'T01064 dse Drosophila sechellia Eukaryotes;Animals;Arthropods;Insects',
'T01065 dsi Drosophila simulans Eukaryotes;Animals;Arthropods;Insects',
'T01067 dwi Drosophila willistoni Eukaryotes;Animals;Arthropods;Insects',
'T01068 dya Drosophila yakuba Eukaryotes;Animals;Arthropods;Insects',
'T01061 dgr Drosophila grimshawi Eukaryotes;Animals;Arthropods;Insects',
'T01062 dmo Drosophila mojavensis Eukaryotes;Animals;Arthropods;Insects',
'T01066 dvi Drosophila virilis Eukaryotes;Animals;Arthropods;Insects']
2.1.3. Look for pathways (by name)
Searching for pathways is quite similar. You can use the find method as above:
>>> print(s.find("pathway", "B+cell"))
path:map04112 Cell cycle - Caulobacter
path:map04662 B cell receptor signaling pathway
path:map05100 Bacterial invasion of epithelial cells
path:map05120 Epithelial cell signaling in Helicobacter pylori infection
path:map05217 Basal cell carcinoma
Note that without the + sign, you get all pathway that contains B or cell. Yet, we have 5 results, which do not neccesseraly fit our request. Alternatively you can use one of BioServices method:
>>> k.lookfor_pathway("B cell")
['path:map04662 B cell receptor signaling pathway']
You can also search for a pathway knowing some gene names but first we need to introspect the pathway to get the genes IDs.
2.1.4. Look for pathway (by genes i.e., IDs or usual name)
Imagine you want to find the pathway that contains ZAP70. As we have seen earlier you can get its gene Id as follows:
>>> s.find("hsa", "zap70")
hsa:7535
The following commands do not help:
>>> s.find("pathway", "zap70")
>>> s.find("pathway", "hsa:7535")
>>> s.find("pathway", "7535")
We provide a method to search for pathways that contain the required gene Id. You can search by KEGG Id or gene name:
>>> res = s.get_pathway_by_gene("7535", "hsa")
>>> s.get_pathway_by_gene("zap70", "hsa")
['path:hsa04064', 'path:hsa04650', 'path:hsa04660', 'path:hsa05340']
This commands first search for the gene Id in the KEGG database and then parse the output to retrieve the pathways.
2.1.5. Introspecting a pathway
Let us focus on one pathway ( path:hsa04660). You can use the get()
command to obtain information about the pathway.
print(s.get("hsa04660"))
The output is a single string where you can recognise different fields such as NAME, GENE, DESCRIPTION and so on. This is quite limited. In BioServices, we provide a convenient parser that converts the output of the previous command into a dictionary:
>>> s = KEGG()
>>> data = s.get("hsa04660")
>>> dict_data = s.parse(data)
>>> print(dict_data['GENE'])
'10000': 'AKT3; v-akt murine thymoma viral oncogene homolog 3 (protein kinase B, gamma) [KO:K04456] [EC:2.7.11.1]',
'10125': 'RASGRP1; RAS guanyl releasing protein 1 (calcium and DAG-regulated) [KO:K04350]',
'1019': 'CDK4; cyclin-dependent kinase 4 [KO:K02089] [EC:2.7.11.22]',
...
This is fine if we just want the name of the genes but what about their relations? Actually, there is an option with the get method where you can specify the output format. In particular you can request the pathway to be returned as a kgml file:
res = s.get("hsa04660", "kgml")
This file can be parsed to extract the relations. We provide a tool to do that:
res = s.parse_kgml_pathway("hsa04660")
The variable returned is a dictionary with 2 keys: “entries” and “relations”.
You can extract the relations as follows:
res['relations']
It is a list of relations, each relation being a dictionary:
>>> res['relations'][0]
{'entry1': u'61',
'entry2': u'63',
'link': u'PPrel',
'name': u'binding/association',
'value': u'---'}
Here entry1 and 2 are Ids. The Ids can be found in
res['entries']
From there you should consult bioservices.kegg.KEGG.parse_kgml_pathway()
and the KEGG document for more information. You may also look at
bioservices.kegg.KEGG.pathway2sif() method that extact only protein-protein
interactions with activation and inhibition links only.
2.1.6. Building a histogram of all relations in human pathways
Scanning all relations of the Human organism takes about 5-10 minutes. You can look at a subset by setting Nmax to a small value (e.g., Nmax=10).
from pylab import *
# extract all relations from all pathways
from bioservices.kegg import KEGG
s = KEGG()
s.organism = "hsa"
# retrieve more than 260 pathways so it takes time
results = [s.parse_kgml_pathway(x) for x in s.pathwayIds]
relations = [x['relations'] for x in results]
hist([len(r) for r in relations], 20)
xlabel('number of relations')
ylabel('\#')
title("number of relations per pathways")
grid(True)
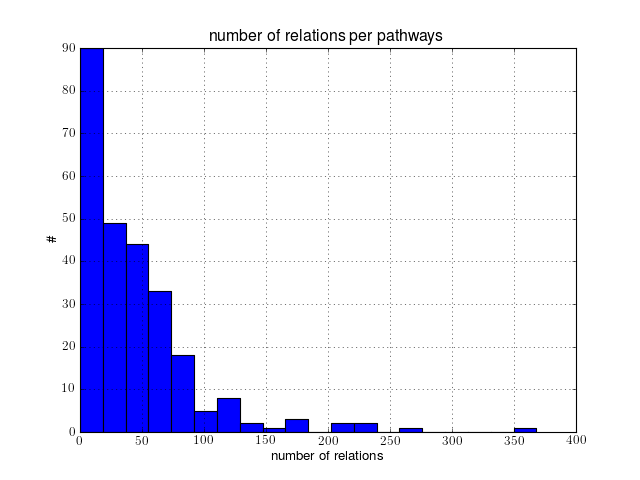
You can then extract more information such as the type of relations:
>>> # scan all relations looking for the type of relations
>>> import collections # for python 2.7.0 and above
>>> # we extract from all pathways, all relations, where we retrieve the type of
>>> # relation (name)
>>> data = list(flatten([[x['name'] for x in rel] for rel in relations]))
>>> counter = collections.Counter(data)
>>> print(counter)
Counter({u'compound': 5235, u'activation': 3265, u'binding/association': 1087,
u'phosphorylation': 940, u'inhibition': 672, u'indirect effect': 559,
u'expression': 542, u'dephosphorylation': 93, u'missing interaction': 80,
u'dissociation': 78, u'ubiquitination': 48, u'state change': 24, u'repression':
12, u'methylation': 2})